
Regulation of protein networks occurs at all levels, from transcription to protein degradation and modification. A key question in mammalian biology has been how the proteome is regulated independently of transcription. With over 600 E3 ubiquitin ligases in human cells, compared to over 1,000 transcription factors, regulation of protein stability is likely on the same scale of significance and complexity as transcriptional regulation. However, protein stability remains an uncharacterized feature for most proteins and this deficiency has arisen because development of tools for a proteome-wide study of protein turnover is technologically challenging. Control of protein turnover serves as a rapid mechanism for the activation or inhibition of signaling pathways when cells respond to environmental changes. Regulated degradation of cancer-related proteins play important roles in cellular transformation, and multiple components of the proteolysis system are directly involved in human diseases. Furthermore, many viruses and bacteria have evolved strategies to hijack the proteolytic pathway of host cells for their own benefit. Therefore, insights from global protein stability profiling are not only critical to further our understanding of biological regulation, but will also provide valuable information for the development of new therapeutic intervention strategies.
Our research aims at developing and employing systematic high throughput genomic and proteomic approaches to the general problem of protein degradation regulation and ubiquitin ligase target identification. Ubiquitin-mediated proteolysis (UPS) is the major proteolytic pathway in the cell and the substrate specificity in UPS is largely conferred by E3 ligases. Many E3s are potential drug targets, but linking an E3 with its substrates is currently a laborious endeavor with few general solutions. We, together with Dr. Stephen Elledge’s lab, developed a method called GPS, for Global Protein Stability, which allows us to simultaneously monitor the stability of ~8,000 proteins in live cells. The method uses a highly parallel multiplex analysis of single cells by FACS coupled with a microarray readout. Many novel findings concerning the mammalian proteome were revealed, such as a strong correlation between protein size and protein stability, and the fact that there are distinct amino acid distributions and cellular functions associated with stable versus unstable proteins. In addition, we applied the GPS approach to identify the substrates of SCF, a member of cullin-RING E3 ligases (CRL). We recovered 75% of the previously known SCF targets and identified several hundred novel substrates. These results demonstrate the potential of GPS as a general platform for the global discovery of E3-substrate networks.
GPS approach opened many adventures for protein turnover studies. GPS profiling could be used to identify proteins whose stabilities change in response to stimuli, genetic perturbations, or during developmental transitions. GPS could also be used to generate disease-specific protein stability signatures that may be useful for diagnoses and elucidating disease mechanisms. GPS could be coupled with loss of function (RNAi), gain of function or chemical screens to discover proteins/compounds that regulate the stability of a protein of interest. Furthermore, the integration of global protein stability information from GPS with other datasets, such as the transcriptome, interactome and phosphoproteome, will provide a global view of regulatory networks and identify crosstalk between protein turnover and other forms of biological regulation.
Our current projects focus on developing the next generation of GPS with improved sensitivity, fidelity, applicability and throughput. We also continue to characterize the function of CRL family E3 ligases and seek to elucidate the mechanism and biology of CRL-mediated protein degradation. In the long term, we are interested in investigating how protein turnover regulation coordinates with other levels of regulation in biological networks, and in developing other high throughput technologies for global analysis of additional forms of protein regulation.
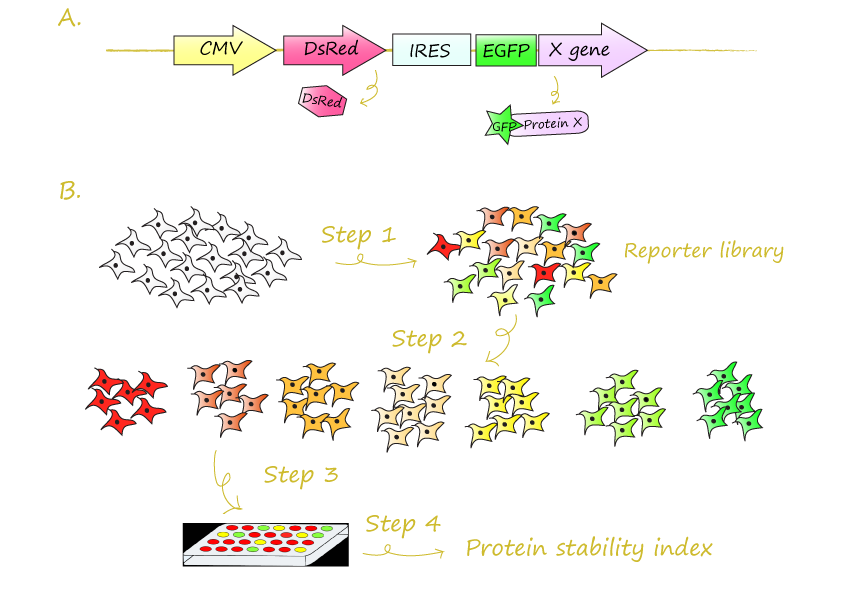
(A) A schematic representation of the GPS reporter construct. The DsRed and GFP-X fusion proteins are both expressed from the same promoter. The tested protein X is fused in frame with the coding sequence of GFP. (B) Schematic diagram of the four steps in GPS profiling
- Step1: Generate GPS reporter cell library. Cells with different GFP/DsRed ratios were painted with different colors.
- Step2: Fractionate reporter cells into subpopulations by FACS according to their GFP/DsRed fluorescence ratios.
- Step3: Recover the GFP-fused cDNAs and quantify by microarray.
- Step4: Derive relative stability of GFP-fused protein from the array result.
Protein Quality Control
Proteome fidelity is of critical importance in almost all cellular processes, yet it is constantly challenged by a diversity of protein aberrations arising from genetic mutations, erroneous transcription and translation, improper folding, and damage induced by various environmental stresses. A protein quality control system must sort through unlimited variations representing “bad” or “good” proteins and selectively eliminate the “bad” proteins. How can such a wide spectrum of defective proteins be captured using limited inspectors? Which molecular characteristics distinguish proteins directed to the “good” pool versus the “bad” pool? Our study addresses these long-standing fundamental questions, which have a broad impact on biology.
Protein termini are indicators of protein integrity. Discovered by Varshavsky et al. in 1986, N-degrons were the first degradation signals to be identified in short-lived proteins. In 2015, we opened a second chapter in “end-mediated” proteolysis by characterizing a C-degron pathway, by which proteins with illegitimate C-termini are eliminated. We uncovered the role of the C-degron pathway in clearing mislocalized proteins, products of proteases, and truncated selenoproteins due to translation errors. Importantly, we have provided evidence supporting that avoidance of C-degron-mediated degradation shapes eukaryotic protein evolution/selection. Accordingly, C-terminal-specific recognition for subsequent protein degradation is likely more prevalent than heretofore acknowledged. Our current research aims to identify additional functions of the C-degron pathway.
Protein Abundance Control
Proteins and their abundances determine the physiological state of a cell. The intracellular concentration of proteins is determined by a balance between the rates of protein synthesis and protein degradation, yet most current studies either consider protein synthesis and degradation separately and treat them as independent events or use RNA abundance as a proxy for protein expression. Experimental evidence suggests that proteolysis-mediated decoupling of transcript and protein abundances provides a post-translational safeguard against the proteomic imbalance caused by genomic mutations or supernumerary chromosomes that may be harmful to the cell. We aim to characterize proteome-wide the interrelationship between protein synthesis and degradation and to investigate the molecular basis and physiological functions of proteolysis-mediated dosage compensation.